paired end sequencing reads
Genes can be regulated by regions far from the. The flexible system enables microbiologists to scale studies from one to hundreds of samples.

Detecting And Characterizing Circular Rnas Rna Seq Blog Segmentation Circular Biochemical
In genetics shotgun sequencing is a method used for sequencing random DNA strands.

. For paired-end data where the sequenced reads are the ends of the same DNA fragment one can use extra information to improve read placement. During paired-end PE chemistry cluster sizes increase slightly due to extra cycles of amplification which can lead to an increase in the number of overlapping clusters. Interpretation of colors is discussed briefy here and in more detail in Interpreting Color by Insert Size and Interpreting Color by Pair Orientation.
It performs both single- and paired-end runs with adjustable read lengths from 1 36 base pairs to 2 300 base pairs. Sanger sequencing uses the SBS approach in which a DNA polymerase generates DNA reads from a template that is the DNA molecule to be analyzed. The default in the submission form is that 10 Ns in a row represent a gap and that paired-ends is the evidence that the sequences on either side of the gap are linked.
With overclustered flow cells this can affect run image registration and lead to poor Q30 scores and possible run failures Figure 2. Sequencing The MiSeq System can deliver 2 300 bp reads and up to 50 million paired-end reads generating up to 15 Gb of data. Where index_prefix is the index for the reference genome generated from bwa index and input_readsfastq input_reads_pair_1fastq input_reads_pair_2fastq are the input files of sequencing data that can be single-end or paired-end respectively.
To run with Bowtie2 execute. Chromatin Interaction Analysis by Paired-End Tag Sequencing ChIA-PET or ChIA-PETS is a technique that incorporates chromatin immunoprecipitation ChIP-based enrichment chromatin proximity ligation Paired-End Tags and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide. The nature of the nucleotide at a given position is now determined using specific dyes.
Micro and nano flow cell options and accompanying reagents are available to support lower-throughput. The chain-termination method of DNA sequencing Sanger sequencing can only be used for short DNA strands of 100 to 1000 base pairsDue to this size limit longer sequences are subdivided. I had the same issue but with Paired End Reads and I solved using samtools and bamToFastq.
This means your two reads are the reverse complement of the 100 3-most bases of the Watson strand and the Crick strand. For example 1 million clusters on a flow cell would produce 1 million single reads and 2 million paired. The Illumina MiSeq system allows a wide-range of sequencing applications and due to its lower data output per run the MiSeq is adequate for small scale projects requiring one to 25 million reads in total or about 510 Mbases to 15 Gbases of raw data.
Paired-end sequencing enables both ends of the DNA fragment to be sequenced. This section covers viewing reads as pairs coloring of mapped paired reads and the split-screen view. A single run can produce output data of up to 15 Gb in as little as 4 h of runtime and can output up to 25 M single reads and 50 M paired-end reads.
In conventional paired-end sequencing you simply sequence using the adapter for one end and then once youre done you start over sequencing using the adapter for the other end. Because the distance between each paired read is known alignment algorithms can use this information to map the reads over repetitive regions more precisely. Typically NISC generates read lengths of 150 bases on a NovaSeq 6000.
For the latter reads are generated from both ends of longer fragmented DNA or RNA. Simple SLURM script for running bwa mem on Crane. It works with either single or paired-end reads and provides a wide range of options appropriate for different sequencing applications.
If this is incorrect then make the appropriate selections for your genome. Fill out metadata on the sequencing and assembly of the genome. For both cases read can be single end or paired ends.
1 shows a schematic view of an Illumina paired-end read. 1A 1B Figure 1. Thus MiSeq provides an ideal platform for rapid turnaround time.
Paired-end reads generate a total of 300 bases of sequence 150b from each end from each fragment in the. In genome sequencing fragmented genomic DNA are sequenced and whole genome is assembled from the reads sequence. We have adopted the latter one for single-end sequencing data.
Additional options for bwa mem can be found in the BWA manual. Namely most of the time except ends spanning CNVs ends should map in proper orientation within a certain distance defined by the average. It is named by analogy with the rapidly expanding quasi-random shot grouping of a shotgun.
This methodology makes it easier to map reads and can be used to improve detection of genomic rearrangements repetitive sequence elements and RNA gene fusions or splice variants. You have two databases one prefixed bact_rrna_db and the other prefixed human_rna_db and your sequence files are seq1fastq and seq2fastq. Whole Exome Sequencing WES is an efficient strategy to selectively sequence the coding regions exons of a genome typically human to discover rare or common.
Instead of single end reads say you have paired end reads and you want to separate the reads that came from bacterial mRNA bacterial rRNA and human RNA. You can find bamToFastq here. Paired-end libraries are created like regular fragment libraries but they have adaptor tags on both ends of the DNA insert that enable sequencing from two directions.
On the other hand RNA-seq tries to sequence reads taken from RNAs. There is a unique adapter sequence on both ends of the paired-end read labeled Read 1 Adapter and Read 2 Adapter. It is considerably faster than existing methods by an order of magnitude for gene-level summarization and requires far less computer memory.
Transcriptome paired-end sequencing reads of 100 bp were generated using a Hiseq 2500 instrument Illumina San Diego CA USA to a targeted depth of 100 million reads per sample. Vernet in Genetics and Evolution of Infectious Diseases Second Edition 2017 21 Sanger Sequencing. These reads are assumed to be identical to.
Sanger sequencing although too. Read 1 often called the forward read extends from the Read 1 Adapter in the 5 3 direction towards Read 2 along the forward DNA strand. MiSeq read lengths can vary between single end reads of 36 bases to paired end runs of 2x300 bases.
IGV provides several features for working with paired-end alignments. If you need unmappedR1fastq containing both paired and unpaired R1 unmapped reads and unmappedR2fastq containing both paired and unpaired R2 unmapped reads Use samtools -f 4 to extract all unmapped reads.

Exon Based Strategy To Identify Differentially Expressed Genes In Rna Seq Experiments Experiments Statistical Analysis Gene

Next Generation Sequencing Has Made It Possible To Perform Differential Gene Expression Studies In Non M Gene Expression Expressions Next Generation Sequencing

Rna Extraction Method Read Length And Sequencing Layout Single End Versus Paired End Contribute Strongly T Interactive Notebooks Method Statistical Analysis

In This Study Researchers From Stanford University Generated Two Paired End Rna Seq Datasets Of Differing Read Lengths 2 7 Rna Sequencing Long Reads Analysis

An Efficient Method For Identifying Gene Fusions By Targeted Rna Sequencing From Fresh Frozen And Ffpe Samp Rna Sequencing Sequencing Chromosomal Abnormalities
Shrna Seq Data Analysis With Edger Rna Seq Blog Analysis Data Analysis Rna Sequencing

Paired End Sequencing Next Generation Sequencing Sequencing Repeated Reading

Exon Based Strategy To Identify Differentially Expressed Genes In Rna Seq Experiments Experiments Statistical Analysis Gene

Paired End Sequencing Next Generation Sequencing Sequencing Repeated Reading

Esnv Detect A Computational System To Identify Expressed Single Nucleotide Variants From Transcriptome Sequencing Data Next Generation Sequencing Sequencing

Here Researchers At The Icahn School Of Medicine At Mount Sinai School Present A New Geometrical Multivariate Appro School Of Medicine Gene Expression Approach
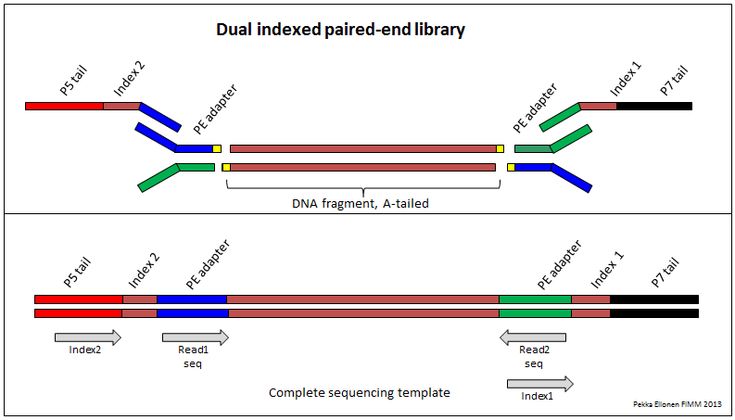
Demultiplexing Dual Indexed Miseq Fastq Files Seqanswers Data Science Index Hypothesis

Diffusion Maps For High Dimensional Single Cell Analysis Of Differentiation Data Analysis Differentiation Cell

Pin On Other Tools

Rna Seq Data Detection Gene

An Efficient Method For Identifying Gene Fusions By Targeted Rna Sequencing From Fresh Frozen And Ffpe Samp Rna Sequencing Sequencing Chromosomal Abnormalities

Funpat Function Based Pattern Analysis On Rna Seq Time Series Data Dynamic Expression Data Nowadays Obtained U Analysis Functional Analysis Rna Sequencing

The Advent Of Next Generation Sequencing And In Particular Rna Sequencing Rna Seq Technologies Has Expande Rna Sequencing Coding Next Generation Sequencing

Methode Sequencage Assemblage Genomique Fonctionnelle Vegetale Enseignement Et Recherche Biochimie Universite Angers Emmanue Biochimie Genomique Enseignement